What is biliary atresia?
Biliary atresia is a liver disease affecting infants. It causes damage, scarring and blockage of the bile ducts. In healthy babies, bile ducts carry bile from the liver to the gallbladder and eventually to the small intestine. Bile includes chemicals that the body is trying to get rid of. When the bile ducts are blocked, bile flow stops. This causes these bad chemicals to collect in the liver. This is called cholestasis. Biliary atresia only occurs in young infants (less than 3-4 months of age). Biliary atresia is a rare disease affecting 1 in 8,000 to 1 in 18,000 live births worldwide. About 10-20% of infants with biliary atresia have abnormalities in other organs, such as heart defects or issues with their spleen.
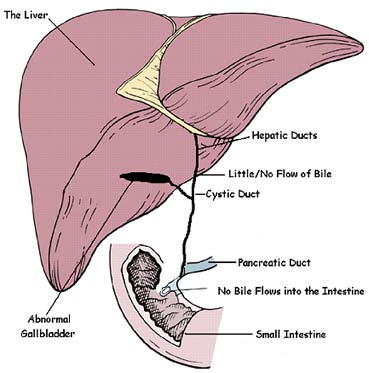
What causes an infant to have biliary atresia?
The cause of biliary atresia is unknown. A number of ideas are being tested. Some doctors think a viral infection can lead to biliary atresia. Others think it may be caused by an overly-active immune system, a problem in development of the bile duct, or a chemical or something toxic that damages the bile duct. Others think a genetic reason or a combination of these things could cause biliary atresia. No matter what causes it, biliary atresia leads to inflammation and scarring of bile ducts outside of the liver and inside the liver. The scarring can lead to cirrhosis (severe scarring) by 6 to 12 months of age. When a liver is badly scarred it cannot do its job correctly. Doctors in the ChiLDReN Network are trying to understand biliary atresia and find better ways to treat it.
What are the symptoms of biliary atresia?
Infants with biliary atresia have jaundice or yellowing of their eyes or skin. Jaundice is normal in many newborns, and should typically clear by 2 weeks of age, particularly in breast fed babies. The jaundice that is part of biliary atresia is caused by a certain substance called “bilirubin” building up in the blood. Any infant with jaundice that lasts past 2 weeks of age should be seen by a doctor. The doctor can check the baby’s blood to see if the liver is working correctly. Infants with biliary atresia may also have stool color that is lighter than normal. This is called “acholic stools” and is the result of bile (which gives stool color) not getting to the intestines. Infants with biliary atresia may also have dark urine, a large liver or a large spleen. If infants have problems in other parts of the body such as heart defects, they can have a heart murmur, low oxygen levels or poor growth. As the liver disease gets worse, other problems might develop, too. Some of these are fluid in the belly (ascites) and itchiness. In healthy babies and children, bile helps with digestion and absorption of food and vitamins. Infants and children who don’t have good bile flow because of biliary atresia may have a hard time gaining weight and absorbing nutrients.
How is biliary atresia diagnosed?
Infants who have jaundice that lasts beyond 2-3 weeks of age or who have light colored stools should be seen by a doctor. The doctor can check the baby’s blood to see if the liver is working correctly. If the doctor thinks the liver is not working correctly, the baby should be checked for biliary atresia. It is very important to diagnose biliary atresia as soon as possible because scarring and cirrhosis in the liver can occur rapidly if the disease is not treated. To help make the diagnosis, the medical team may order a number of tests, including 1) blood and urine tests looking for other common or treatable causes of jaundice, 2) an ultrasound to look at the bile ducts, gall bladder and liver, 3) a liver biopsy, or 4) a HIDA scan (hepatic scintigraphy) to see if bile is getting into the intestine. If these tests point to a possible diagnosis of biliary atresia, your doctor may order the intraoperative cholangiogram, a surgical operation where the bile ducts are searched for and then dye is injected directly into the gallbladder to get the best look possible at the bile ducts. If bile ducts cannot be seen by this testing, then the surgical operation is continued to treat the biliary atresia by the Kasai procedure.
How is biliary atresia treated?
If the testing and cholangiogram show biliary atresia, infants need a surgical operation for treatment, known as the Kasai procedure or hepatoportoenterostomy. In this procedure, the blocked bile ducts are removed and a piece of the small intestine is directly connected to the liver in place of the bile duct. This allows bile to leave the liver and enter the intestine. Other supportive medical treatments are recommended before and after the Kasai procedure. These include making sure nutrition is ideal by using infant formulas with higher calories or certain types of fat. Doctors might also use additional vitamins, ursodeoxycholic acid, and low-dose antibiotics to prevent bacterial infection of the bile ducts (cholangitis). When the Kasai procedure is successful, the stools become yellow or brown, the bilirubin normalizes in the blood and jaundice disappears, and nutrition and well being improve.
Steroids (e.g., prednisone) have been used in the past to try to decrease inflammation and improve bile flow after the Kasai procedure. The ChiLDReN network recently completed a placebo-controlled trial of steroids in infants with biliary atresia after the Kasai procedure. The investigators found that steroids do not make any difference in how well bile flows after a Kasai procedure, and do not recommend routine steroid treatment.
The ChiLDReN study is currently conducting a clinical trial of IVIg (intravenous immunoglobulin) after the Kasai procedure to see if it is safe to use and if it changes the course of this disease by treating the inflammation and the activated immune system.
What is the outlook for infants diagnosed with biliary atresia?
The chance of a successful Kasai procedure is best if the procedure is done before 30-45 days of age. Occasionally, infants who are not diagnosed until after 3-4 months of age and already have cirrhosis at the time of diagnosis, do not have a Kasai procedure. These infants are then listed for liver transplantation without undergoing a Kasai.
If the Kasai procedure is successful, jaundice disappears and bilirubin levels return to normal. This happens in 40-60% of infants having a Kasai procedure. Even with the Kasai procedure, many children with biliary atresia still develop more scarring in the liver and eventually need a liver transplant.
Biliary atresia is the most common reason for liver transplantation in children. About half of infants who undergo a Kasai procedure will still need liver transplantation by three years of age. In about one quarter of infants, the jaundice will go away at first, but those children will need liver transplantation by the teenage years because of slowly progressive liver scarring. With our current therapies, only about one quarter of babies who have the Kasai procedure will survive to their 20's without needing liver transplantation.
Although liver transplantation is a major surgical procedure requiring long term immunosuppression medicines, this life-saving procedure has markedly improved survival rates of infants and children with biliary atresia. Long-term survival after liver transplantation for biliary atresia is over 90%.
How can I learn more about biliary atresia?
The LearnAboutBA app is now available for both iTunes and Android users. This new app (designed by the team at The Hospital for Sick Children in Toronto) aims to support the teaching of patients, families and healthcare providers about this important liver disease. Download it for free from the iTunes App Store or the Google Play store.
Does the ChiLDReN Network have any studies that include patients with biliary atresia?
Yes. The ChiLDReN Network has several studies that include patients with biliary atresia.
The PROBE, BASIC, and FORCE studies are natural history studies that include patients with biliary atresia. A natural history study is aimed at acquiring information and data that will provide a better understanding of rare conditions. Participants will be asked to allow study personnel to obtain information from medical records and an interview, and to collect blood, urine, and tissue samples when clinically indicated, in order to understand the causes of these diseases and to improve the diagnosis and treatment of children with these diseases. All of the information obtained in these studies is confidential and no names or identifying information are used in the study.
PROBE: A prospective study of infants and children with cholestasis.
Eligibility: Infants up to 6 months of age that have been diagnosed with cholestasis (direct hyperbilirubinemia).
ClinicalTrials.gov Study NCT00061828
BASIC: A prospective database study of older children with biliary atresia.
Eligibility: Children and adults age 6 months and older that have been diagnosed with biliary atresia, both before and after liver transplantation.
ClinicalTrials.gov Study NCT00345553
FORCE: A cross-sectional and longitudinal assessment of the utility of liver stiffness measurement (as assessed by elastography) in children with chronic cholestatic liver disease.
Eligibility: Children currently enrolled in BASIC, PROBE, or LOGIC with a diagnosis of biliary atresia, alpha-1 antitrypsin deficiency or Alagille syndrome.
ClinicalTrials.gov Study NCT02922751
Are there any organizations or foundations that help families dealing with biliary atresia?
Yes. The ChiLDReN Network works with numerous groups that support patients and families who are dealing with rare liver diseases. Please click here to go to that page on our website (Support Groups). You will see the list of groups and information about them.