Definition
Alagille syndrome (ALGS) is a dominantly inherited multisystem disorder consisting of abnormalities of the liver, heart, eye, spine, facies, kidney, vasculature and other organs. It is primarily caused by mutations in JAGGED1, a ligand in the Notch signaling pathway. A minority of patients in whom a JAGGED1 mutation could not be identified has been found to have mutations in Notch2, a receptor in the Notch signaling pathway. Notch genes control cell fate determination and are active during (and sometimes after) fetal development. Mutations in Notch ligands and receptors have recently been demonstrated to cause several disorders of embryogenesis.
Dr. Daniel Alagille, a French Pediatric Gastroenterologist, first described a syndrome consisting of characteristic facies, intrahepatic bile duct paucity and cholestasis, heart murmur, butterfly vertebrae and posterior embryotoxon in 1969. Since that original description, abnormalities in other organs have been recognized to be common manifestations in ALGS, namely the kidneys and vasculature.
Etiology
ALGS is one cause of neonatal cholestasis, with a minimum incidence of approximately 1:30,000 live births. It is inherited in autosomal dominant fashion, but approximately 60% of cases are caused by de novo mutations. ALGS is a genetically heterogenous disorder, caused by mutations in the Notch signaling pathway ligand gene, JAGGED1, or the gene for its receptor, NOTCH2. At this time the vast majority of patients have mutations in JAGGED1 (94%) with only a small subset (approximately 2%) having mutations in NOTCH2. Notch signaling is triggered when a ligand on the surface of one cell communicates with a Notch receptor on the surface of an adjacent cell. The end result of Notch signaling affects cell fate determination. Notch was first discovered in Drosophila, where mutations cause a characteristic defect or notching in the wing. Notch and its ligands (Jagged, Delta and Serrate) are active embryologically in all of the organs affected in ALGS. Mutations in members of the Notch signaling pathway have been shown to cause human disease. Mutations in NOTCH1 have been found to cause aortic valve disease, mutations in NOTCH3 cause CADASIL (cerebral autosomal dominant arteriopathy and stroke), and mutations in the Notch ligand delta-like3 (Dll3) cause the autosomal recessive disorder spondylocostal dysostosis.
The location of the causative gene was first suggested by case reports of ALGS associated with cytogenetically visible deletions of the long arm of chromosome 20. In 1997, mutations in JAGGED1 were shown to be the cause of ALGS. Mutations are now demonstrated in approximately 95% of patients with clinical ALGS. The majority of the mutations detected result in protein truncation, and haploinsufficiency is the presumed mechanism of pathogenesis. The mutations are distributed widely over the entire coding region, and most are unique. Genetic testing is fairly straightforward in most cases with the use of readily available cholestasis genetic panels which use next generation sequencing. There are no apparent genotype-phenotype correlations, even for patients with total gene deletion. There is extreme variability of phenotype, even within families, suggesting that other modifying genes or environmental factors contribute significantly to the manifestations of the disease.
Prenatal testing for JAGGED1 mutations is available and accurate if the mutation has previously been identified in the proband. Pre-implantation genetic diagnosis has been successfully performed. The molecular testing has aided in the diagnosis of ALGS for patients with only minor or atypical manifestations. It has expanded the phenotype to include a pedigree with only cardiac manifestations and one with deafness as a major feature. Finally, a study of relatives of probands has shown that the majority of patients with JAGGED1 mutations have less severe disease or only minor manifestations that would not meet the classical diagnostic criteria of Alagille, but which obviously might result in progeny with severe or fatal manifestations of the syndrome. Further testing for mutations of JAGGED1 in other related disorders is likely to further expand the phenotypes associated with this gene and the Notch signaling pathway.
Clinical Features
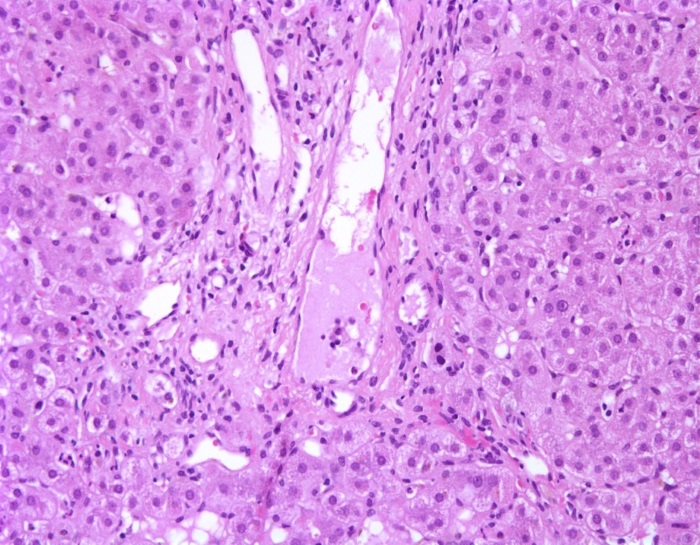
Hepatic features: Intrahepatic interlobular bile duct paucity is one of the most common features of ALGS, however it may not be present in many infants younger than 6 months of age and may not be present in many mildly affected individuals. Bile duct paucity is defined as a ratio of bile ducts to portal tracts of less than 0.5 in a liver biopsy with an adequate (≥10) number of portal tracts present (Figure 1). The normal number of bile ducts in a portal tract increases throughout the first years of life, reaching a normal ratio of nearly 2 by adolescence. The normal ratio in premature infants is less than 1, so care must be taken in this group when interpreting liver histology.
FIGURE 1:
Liver specimen from an infant with Alagille syndrome with bile duct paucity. The portal tract is shown without any identifiable interlobular bile duct. (Hematoxylin and eosin staining, 200x magnification)
Many infants present with cholestatic jaundice in the first days or months of life. Typically, the total and conjugated or direct bilirubin and GGT are quite elevated. Discrimination from biliary atresia is very important, but can be difficult, particularly from the syndromic type of biliary atresia, which is associated with other anomalies such as congenital cardiac disease. In general, a percutaneous liver biopsy may discriminate biliary atresia (with bile duct proliferation) from ALGS (with bile duct paucity), but in younger infants there is considerable overlap of the histologic pattern with bile duct proliferation being common in ALGS infants less than 6 months of age. In biliary atresia, an operative cholangiogram will not demonstrate continuity of the ducts from the intrahepatic to distal extrahepatic tree. Unfortunately, the intrahepatic tree in ALGS can be extremely hypoplastic, and apparent (but incorrect) non-opacification of the proximal ducts has resulted in a significant number of ALGS patients being misdiagnosed as biliary atresia, and therefore receiving a Kasai hepatoportoenterostomy. It appears that this operation may actually adversely change the course of ALGS and increase the likelihood of liver transplantation.
The severity of cholestasis in ALGS patients is highly variable. Nevertheless, patients with ALGS may have very significant cholestasis compared to other pediatric liver diseases. Typically the cholestasis worsens throughout the first months or years of life, and then in some patients there is a subsequent gradual stabilization or improvement. A total bilirubin between 5-10mg/dL between 6-12 months of age has recently been shown to be associated with an almost 5-fold increased risk of liver transplantation. Peak values can include total bilirubin > 20 mg/dl, GGT > 2,000 U/L, cholesterol > 2,000 mg/dl and bile acid levels >100 times normal. Hepatic synthetic function is usually well maintained. In infants and children, xanthomas are common, and can be quite extensive. Pruritus can be debilitating, despite therapy with ursodeoxycholic acid, hydroxyzine, and rifampin. Intestinal bile acid transporter (IBAT) inhibitors have recently been approved for use in children with ALGS and cholestatic pruritus. This class of drugs interrupts enterohepatic circulation of bile salts by bocking their intestinal uptake. Clinical trials performed in patients with ALGS and pruritus have demonstrated efficacy in reducing pruritus, serum bile acids and improving measures of sleep and quality of life. With the advent of these therapies, fewer children are being offered surgical biliary diversion.
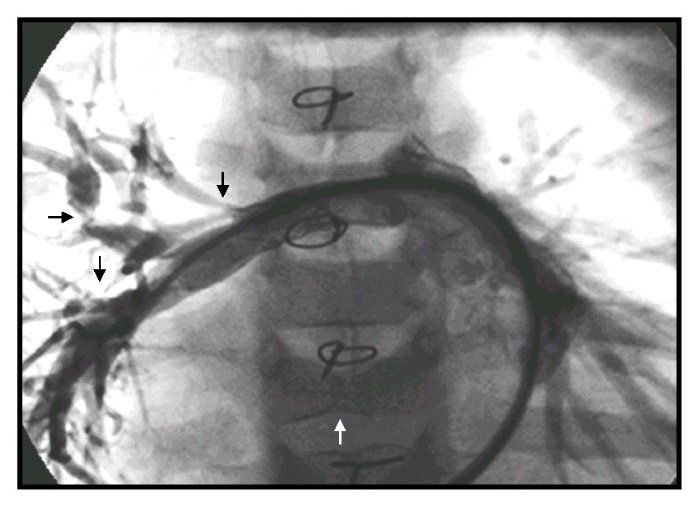
Cardiopulmonary features: The majority of murmurs in ALGS are caused by peripheral pulmonary artery stenosis (PPS) (Figure 2) rather than an intracardiac or valvular process. The PPS can be severe and can cause highly disparate perfusion of the right versus left lung. Most patients, however, have trivial PPS. About 7-11% of ALGS patients have significant structural cardiac disease, usually arising in the right heart embryologically. The most important lesion is tetralogy of Fallot, which is sometimes accompanied by pulmonary atresia. Large series of ALGS cardiac lesions have been described. Correction of these lesions can be complicated by the significant pulmonary artery stenoses that increase resistance to right heart flow. The presence and type of intracardiac lesions are major factors in infancy that predict excess mortality in ALGS.
FIGURE 2:
Right pulmonary arteriogram demonstrating multiple stenoses (black arrows) in an Alagille syndrome patient with prior surgery for tetralogy of Fallot, peripheral pulmonic stenoses, and a butterfly vertebra (white arrow).
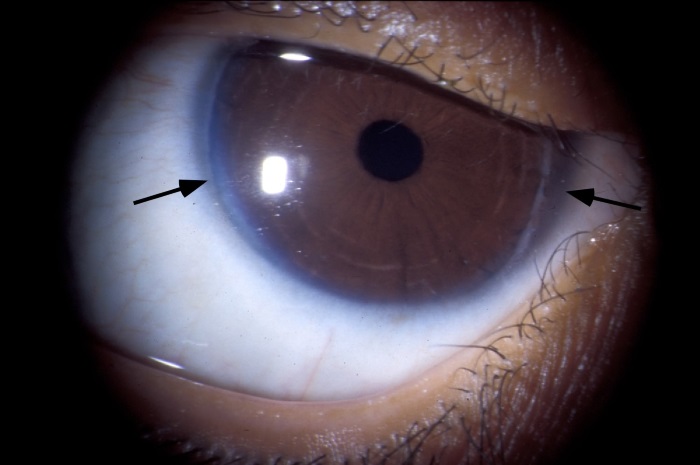
Ocular abnormalities: A large number of ocular manifestations may occur in ALGS. Posterior embryotoxon is the most important diagnostically. Posterior embryotoxon is a prominent, centrally positioned Schwalbe’s line (Figure 3). It generally does not affect vision. It occurs in many (56-95% of different series) ALGS patients but has also been reported to occur in 20% of normal eyes. Axenfeld anomaly (iris strands) is seen in 13% of ALGS patients.
FIGURE 3:
Posterior embryotoxon and prominent Schwalbe’s line (arrows).
Facial abnormalities: The typical facies in childhood consist of a prominent forehead, deep set eyes with moderate hypertelorism, a small pointed chin, and a saddle or straight nose. This has been termed triangular facies (Figure 4). The typical facies in adults with ALGS do not resemble childhood features. The forehead becomes less prominent, and the chin is more protuberant.
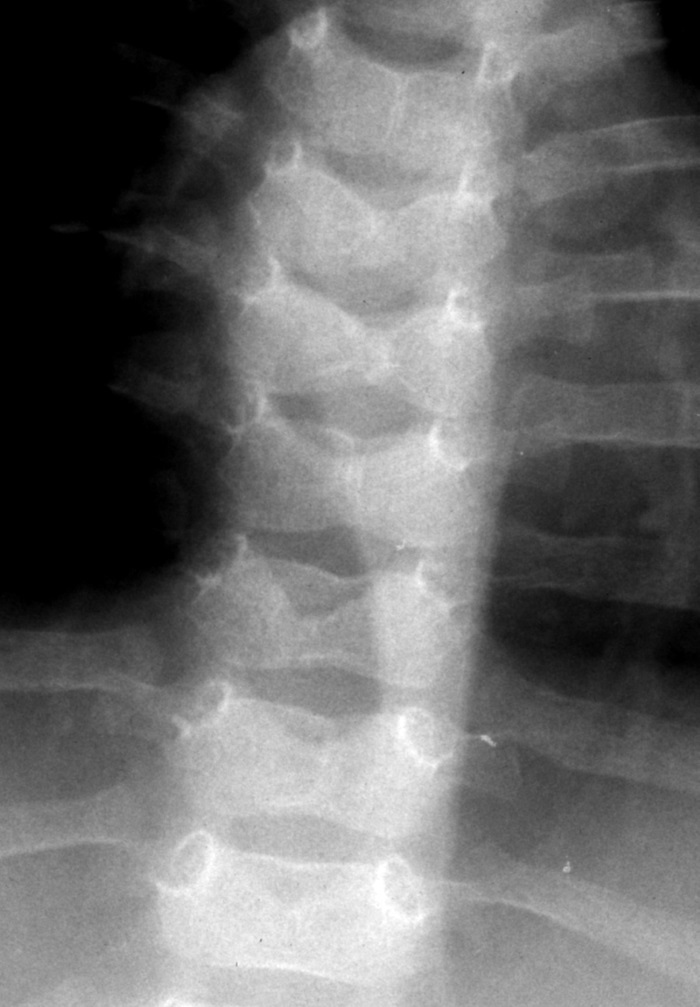
Skeletal abnormalities: The characteristic manifestation of ALGS is the sagittal cleft or butterfly vertebrae, which is found in 33-87% of patients (Figure 5). Due to a failure of the fusion of the anterior arches of the vertebrae, the vertebral body is split sagittally into paired hemivertebrae. This anomaly may occur in other syndromes and also in healthy individuals. It is generally not of structural significance.
FIGURE 5:
Butterfly vertebrae of T5 and T6, with vertebral anomalies at T4, T7, T8 and T9 in an infant with Alagille syndrome.
Severe metabolic bone disease with pathologic fracturing is common in ALGS. Factors that may contribute to osteopenia include chronic malnutrition, deficiencies of vitamins D and K, chronic hepatic and renal disease, mineral deficiency and pancreatic insufficiency. In addition to these factors, the increased frequency of long bone fractures that is seen in ALGS has led to the suggestion that there may also be an intrinsic Notch-mediated defect in bone.
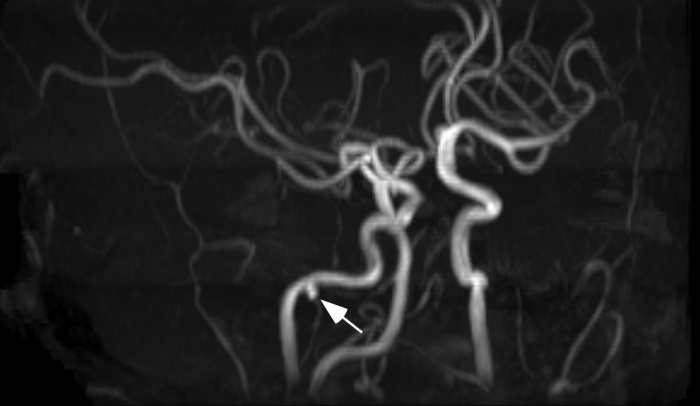
Central Nervous System abnormalities: Stroke, intracranial bleeding and other CNS vascular anomalies occur in patients with ALGS, and have contributed significantly to morbidity and mortality. Intracranial bleeding has occurred in up to 15% of cases presenting with liver disease in some series, and is fatal in approximately one third of events. The majority of this bleeding has occurred in the absence of a significant coagulopathy. Head trauma, generally of a minor nature, has been associated with a minority of these bleeding events. Structural vascular lesions, including aneurysms and moyamoya, have been identified in some patients with ALGS (Figure 6).
FIGURE 6:
Aneurysm of the external carotid artery in an adolescent with Alagille syndrome.
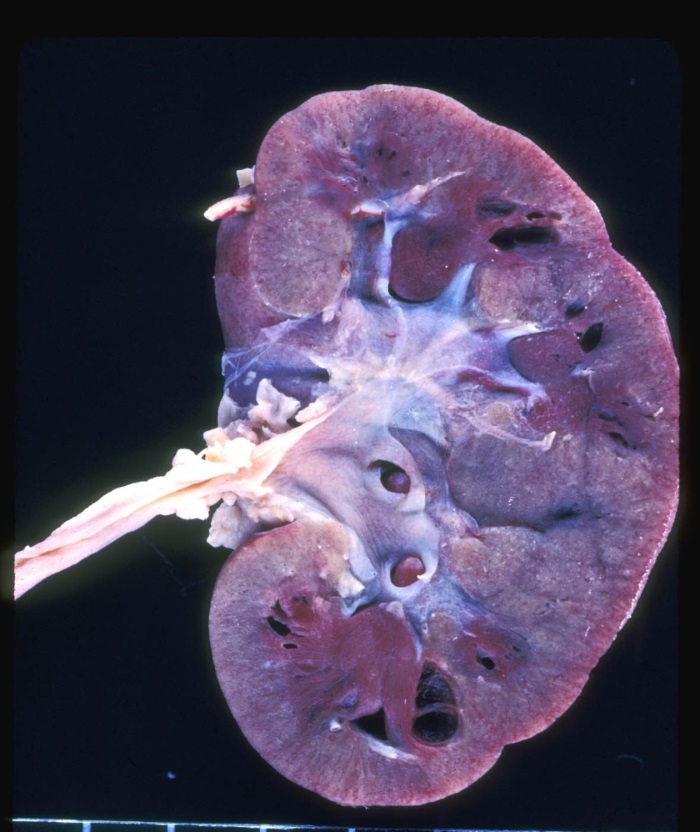
Renal abnormalities: It is now recognized that renal abnormalities are an important feature of ALGS. Major structural disease can occur, including cystic and dysplastic kidneys, solitary kidney, and duplicated ureters (Figure 7). Renal artery stenosis is a cause of systemic hypertension. Tubular disease, including renal tubular acidosis, tubulointerstitial nephropathy, and glomerular lipidosis has been seen in ALGS. Renal transplantation has been described in ALGS. ALGS individuals undergoing liver transplantation are generally considered at increased risk of nephrotoxicity from immune suppressants and should be managed with renal-sparing protocols.
FIGURE 7:
Diffuse renal cystic dysplasia in a patient with Alagille syndrome.
Diagnosis
The diagnosis of ALGS was traditionally made with the combination of bile duct paucity in association with at least three of seven major criteria: cholestasis, heart murmur, embryotoxon, butterfly vertebrae, facies, renal and vascular anomalies. The identification of JAGGED1 mutations and the availability of molecular testing have greatly improved the understanding about and diagnosis of ALGS. It has confirmed the clinical impression that nearly all minimally affected relatives who manifest one or two minor features of ALGS will carry the gene mutation. Since the transmission risk for their progeny is high, it seems appropriate to designate these mutation positive individuals as ALGS. Some mutation positive individuals will have only one or even no clinical features yet may have severely affected progeny.
Treatment
Treatment of ALGS is based on improving bile flow using ursodeoxycholic acid (UDCA; 10-20 mg/kg/day), maintaining optimal nutrition, preventing fat-soluble vitamin deficiencies, addressing pruritus, and treatment of any extrahepatic features. The management of severe pruritus has been modified recently by the approval of IBAT inhibitors which are efficacious in reducing serum bile acids and improving pruritus.
Liver transplantation is effective therapy for patients with hepatic ALGS and is required in 50-75%. Indications for liver transplantation include intractable pruritus, profound fat-soluble vitamin deficiency, severe bone disease (hepatic osteodystrophy), growth failure or complications of portal hypertension. Following transplantation, jaundice, xanthomas and pruritus resolve. Bone density may significantly improve post-transplant, however, growth failure may not. Living related donor selection must be done with caution, as 40% of parents will carry the JAGGED1 mutation, and, even if asymptomatic, may have severely hypoplastic bile ducts that make segmental live-donor donation impossible. Patients with ALGS may have significant renal disease, cardiac disease and malnutrition, each of which may contribute to increased morbidity or mortality of transplantation. Multiple organ transplantation has been successfully performed occasionally in ALGS.
Prognosis
In general, survival with ALGS is very good with almost 90% survival to adulthood. Most of the mortality arises from severe cardiac, liver and vascular issues. Outcomes from liver transplantation are also excellent and indistinguishable from biliary atresia in the modern era, albeit with careful selection and optimization of recipients.
ChiLDReN Network studies that include patients with Alagille Syndrome
The ChiLDReN Network has one study that includes patients with Alagille Syndrome.
The LOGIC study is a natural history study that includes patients with Alagille Syndrome and three other rare liver diseases. A natural history study is aimed at acquiring information and data that will provide a better understanding of rare conditions. Participants will be asked to allow study personnel to obtain information from medical records and an interview, and to collect blood, in order to understand the causes of these diseases and to improve the diagnosis and treatment of children with these diseases. All of the information obtained in these studies is confidential and no names or identifying information are used in the study.
LOGIC: A longitudinal study of genetic causes of intrahepatic cholestasis.
Eligibility: Children and adults ages 6 months through 25 years diagnosed with Alagille Syndrome, alpha-1 antitrypsin deficiency, progressive familial intrahepatic cholestasis, or bile acid synthesis defects.
ClinicalTrials.gov Study NCT00571272
Organizations or foundations that help families dealing with Alagille Syndrome
The ChiLDReN Network works with numerous groups that support patients and families who are dealing with rare liver diseases. Please click here to go to that page on our website (Information for Families). You will see the list of groups and information about them.