Edited by Kevin E. Bove MD
ALAGILLE SYNDROME
The liver disease in Alagille syndrome is characterized by a deficit of bile ducts in portal areas that is progressive and may eventually involve large as well as small caliber bile ducts. The mechanism of duct deficiency, called paucity, is not understood, but probably involves developmental failure as well as progressive destruction. Significant inflammation and non-uniform liver fibrosis are common features in liver biopsy specimens.
Alagille Syndrome: Sequence of the basic pathological lesion in liver of patients
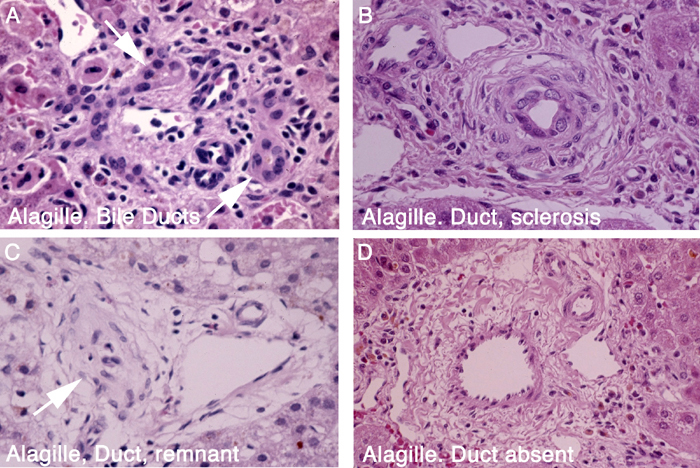
FIGURE 1. The panels in the illustration are from liver biopsy specimens in infants and are arranged to suggest that destruction of small bile ducts is a progressive process that injures and obliterates small interlobular bile ducts, resulting in obstructive cholestasis.
ALPHA-1 ANTITRYPSIN DEFICIENCY
AAT deficiency is most often associated with the Z mutation, which results in abnormal folding of the mutated protein with abnormal accumulation in the endoplasmic reticulum of hepatocytes where proteins are synthesized. Intra-hepatocyte retention of the abnormal AAT protein impairs secretion and results in reduced levels of AAT in the blood. For reasons that are unclear, accumulation of abnormally folded AAT protein in hepatocytes increases risk for different clinical presentations such as transient neonatal cholestasis, persistent liver disease and end-stage liver disease. Moreover, only a minority of children who carry the mutation develop any liver disease. The abnormal protein is recognizable in liver tissue samples but the disease may be diagnosed by blood tests alone.
Alpha-1 Antitrypsin Deficiency: Diagnostic images

FIGURE 1. A: Diatase-resistant inclusions are prevalent in hepatocytes but rare in carcinomatous nodule (*). (PAS+ stain after diatase treatment). B and C: Storage material in hepatocytes is demonstrable by peroxidase immunostain in paraffin section and fluorescein-conjugated antibody in frozen section. D: Alpha-1 Antitrypsin protein accumulates in endoplasmic reticulum of hepatocytes. (Electron Microscopy)
Alpha-1 Antitrypsin Deficiency: 5 week old infant
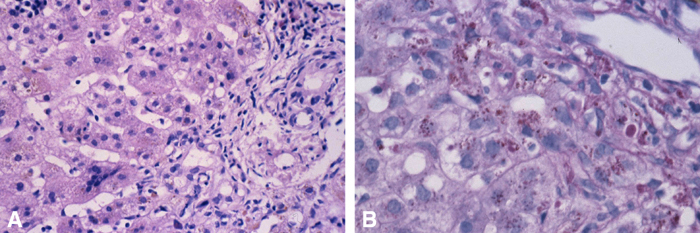
FIGURE 2. A: Neonatal hepatitis with giant cell transformation and focal cholangiopathy. B: Storage globules are readily demonstrated; this is not always the case in affected young infants. (PAS- Diastase stain);
Alpha-1 Antitrypsin Deficiency: Cholangiopathy in Infants
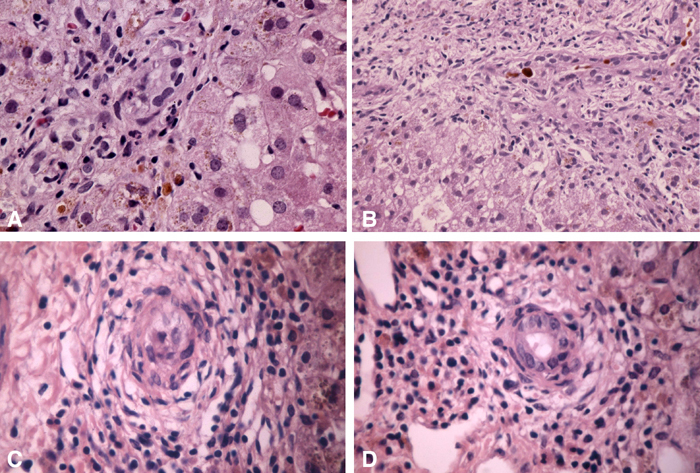
FIGURE 3. A: Mild pericholangitis. B: Ductular cholestasis. C and D: Destructive cholangitis.
Alpha-1 Antitrypsin Deficiency: Cirrhosis in child

FIGURE 4. Cirrhosis in 13 month old child with ZZ phenotype
BILE ACID SYNTHESIS AND METABOLISM DEFECTS
Bile acid defects (BAD). Bile acids are synthesized in the liver from cholesterol and secreted into the bile enters the GI tract and facilitates absorption of lipids and fat soluble vitamins. Multiple known defects in the synthetic pathway result in bile acid deficiency and accumulation of intermediate products of variable toxicity that cause liver damage. Depending on the particular defect, liver damage may be severe and life-threatening with neonatal onset, or mild and chronic. The liver disease and fat soluble vitamin deficiencies may be reversed by replacement therapy with normal bile acids.
Pathways for Bile Acid Synthesis
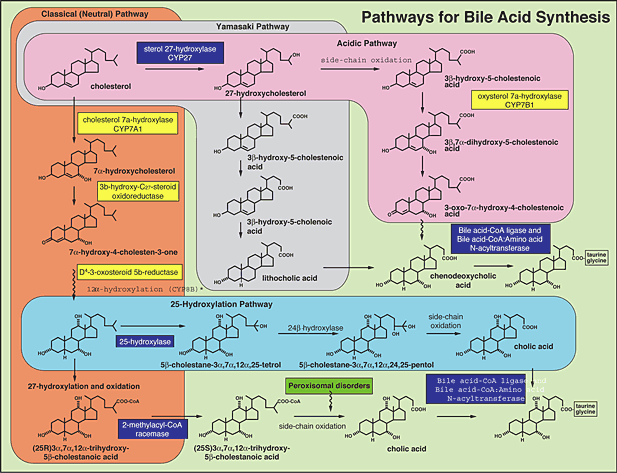
FIGURE 1. Yellow, steroid nuclear defects. Blue, Side chain defects. Green, Peroxisomal disorders.
Bile Acid Synthesis Defect: 3-beta-OH-steroid dehydrogenase deficiency
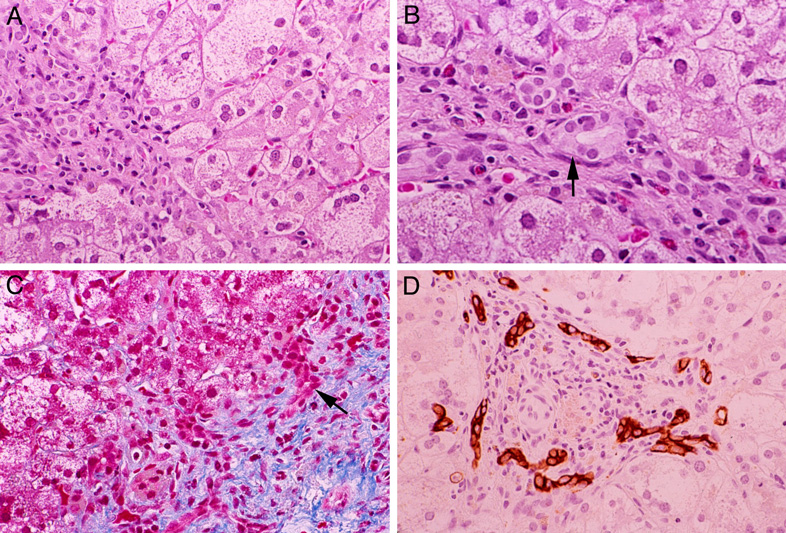
FIGURE 2. In an infant, pretreatment. A: Early onset (age 6 months). Intrabular cholestasis with prominent giant cell transformation. B. Mild portal inflammation and mild swelling of bile duct epitheium. C: Ductular reaction with mild periportal fibrosis. Trichrome stain. D: Ductular reacton is highlighted by cytokeratin stain.
Bile Acid Synthesis Defect: 5 beta reductase deficiency
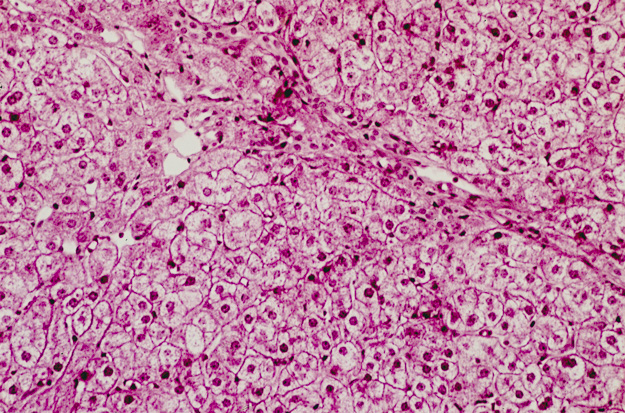
FIGURE 3. Neonatal onset. Oral bile acid replacement therapy for 16 months. Lobular cholestasis resolved. Minimal portal fibrosis remains.
Bile Acid Synthesis Defect: 5 oxysterol beta reductase deficiency
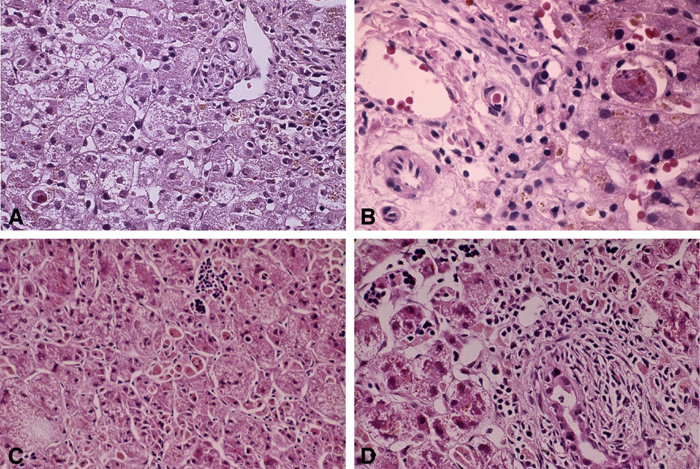
FIGURE 4. In two infants, pretreatment. A and B: Liver biopsy at age 5 days. C: Liver biopsy at age 6 weeks.
Bile Acid Synthesis Defect: 3-beta-OH steroid dehydrogenase deficiency
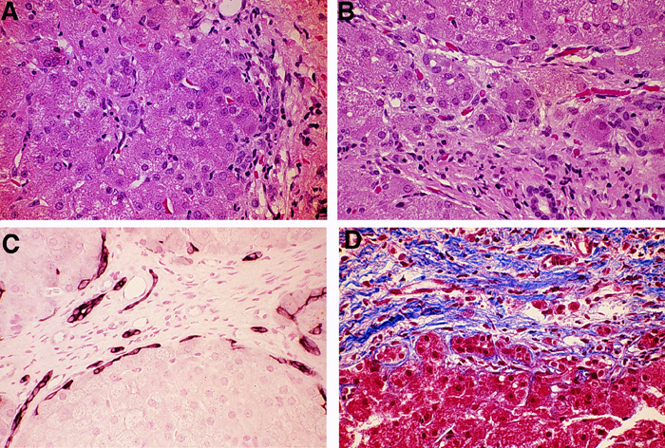
FIGURE 5. Late onset type. All panels show evidence of chronic low-grade ductular reaction accompanied by fibrosis.
BILIARY ATRESIA
Biliary atresia is a fibro-obliterative disorder of the extrahepatic bile ducts that presents in the first weeks of life. The cause is not known but might include genetic, immunologic, toxic and infectious etiologies. Histologic evidence of bile duct obstruction and resultant fibrosis is seen.
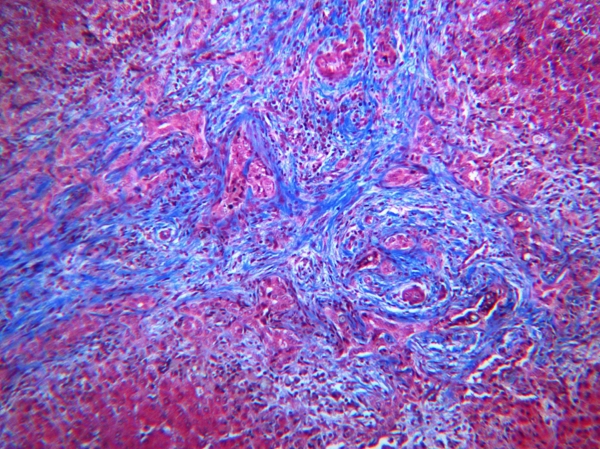
FIGURE 1. This is a microscopic section of liver tissue obtained by needle biopsy in an infant boy at age 45 days with jaundice that had appeared at 2 weeks before. The image shows an enlarged portal area distorted by marked increase in bile duct profiles representing a proliferative response to obstruction of large extra hepatic ducts. Duct proliferation is accompanied by prominent fibrosis that stains dark blue.
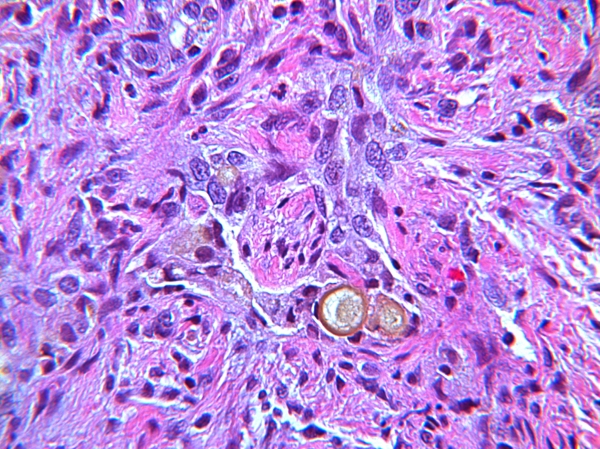
FIGURE 2. Higher magnification of obstructed bile ducts shows a dense plug of bile in one duct profile.
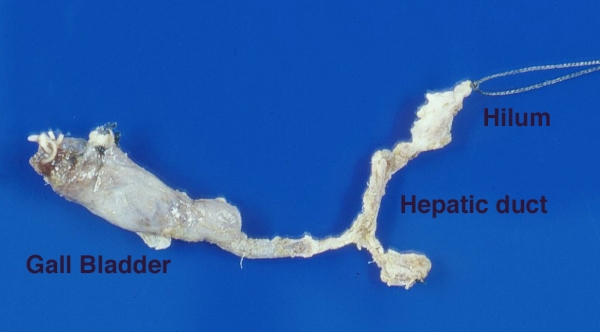
FIGURE 3. Biliary atresia was confirmed at surgery several days later. This is a photograph of the constricted obstructed atretic extrahepatic bile ducts and gall bladder that were excised. A hepatoportalenterostomy (Kasai anastomosis) was constructed in the hope of re-establishing drainage of bile from the liver.
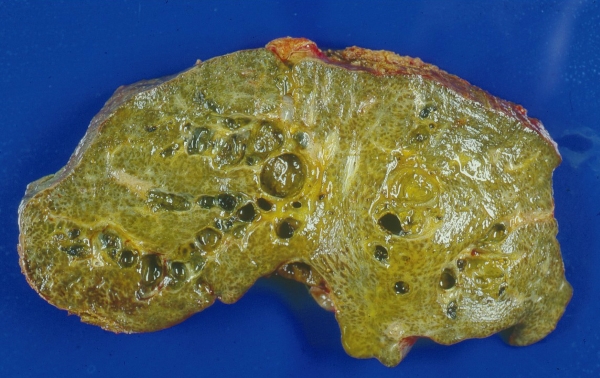
FIGURE 4. The attempt to establish drainage failed in this case (the success rate is about 50%) and 9 months later the liver was explanted and replaced by a donor liver (allograft). This is a cross section of the diffusely fibrotic explanted liver that shows typical advanced biliary cirrhosis.
CYSTIC FIBROSIS LIVER DISEASE
Cystic fibrosis (CF) is a condition cause by a mutation in the CFTR (cystic fibrosis transmembrane regulator), which causes abnormal chloride transport into the lungs, bile ducts, pancreas, and intestine. Neonatal cholestasis or later cirrhosis and portal hypertension can occur.
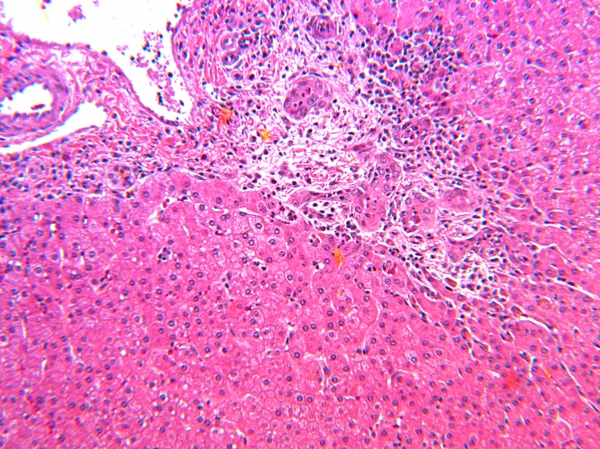
FIGURE 1. Proliferating damaged small bile ducts in this portal area are the seat of acute cholangitis, probably due to ascending bacterial infection superimposed on focal duct obstruction due to cystic fibrosis. Acute inflammation with stromal edema is limited to the portal zone but bile stasis is present in the nearby hepatocytes. X100. H&E stain.
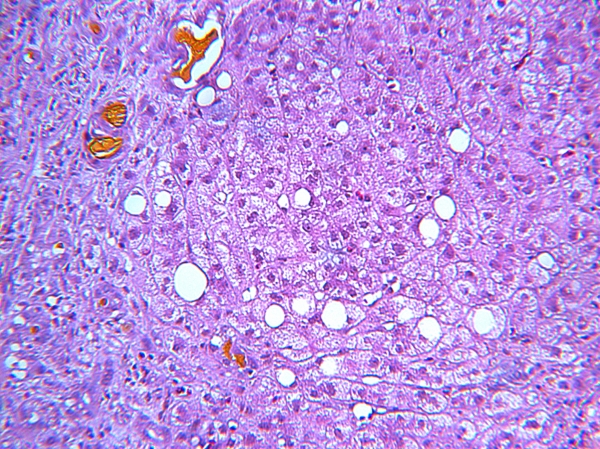
FIGURE 2. Focal intralobular canalicular bile stasis and focal fatty change within scattered hepatocytes result from bile duct obstruction and nutritional disturbance in cystic fibrosis. X100. H&E stain.
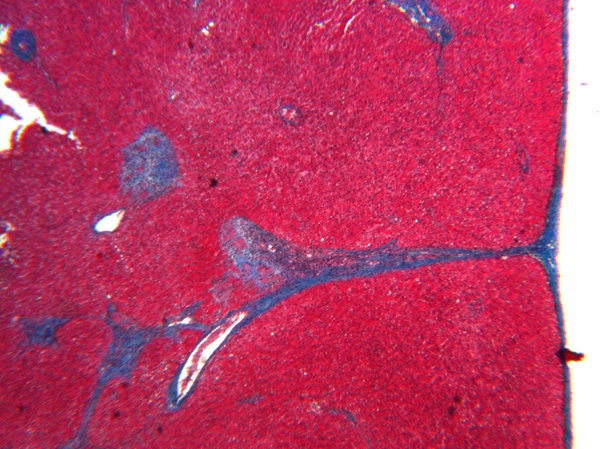
FIGURE 3. Focal delicate scars around portal areas, as shown here, are presumably caused by previous episodes of focal cholangitis. Such scars are common in cystic fibrosis but do not usually cause liver dysfunction. X25, Trichrome stain (Blue-staining scars must be distinguished from normal structural collagen).
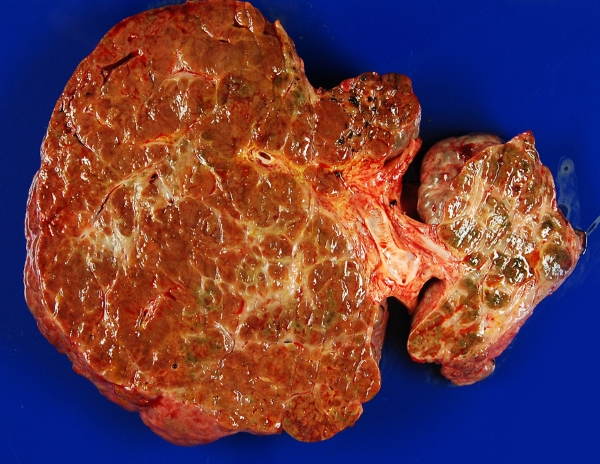
FIGURE 4. Cross sectioned slice of entire liver removed from a teenager with cystic fibrosis who had severe portal hypertension. Extensive but non-uniform streaks of pale scar tissue tend to be more prominent in the central part of this slice near the major bile ducts. This patient received a liver transplant.
MITOCHONDRIAL HEPATOPATHIES
Mitochondrial hepatopathies may involve the liver alone or in concert with other organs such as skeletal muscle, heart, GI tract and brain. These are disorders of energy metabolism that may produce acute liver failure, chronic liver disease or multi-organ dysfunction with or without liver involvement. The morphological changes in the liver may be distinctive at the time of liver failure in infants, but the full spectrum of defects and morphologies is not yet clear.
Hepatic mDNA depletion: Mitochondrial
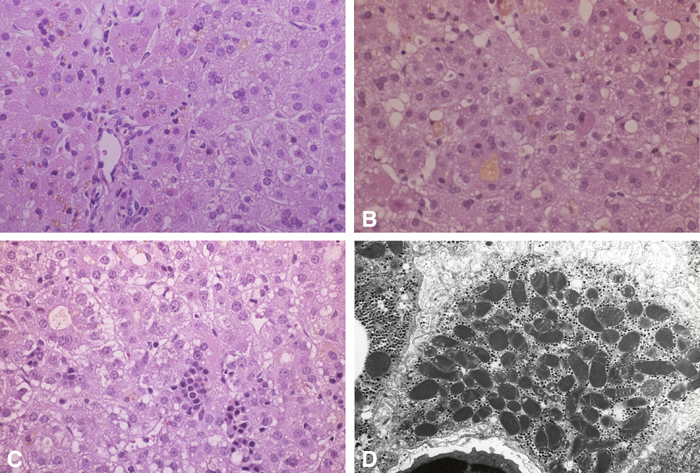
FIGURE 1. Acute liver failure in infancy due to mDNA depletion: Notable are non-uniform microvescicular fat, canalicular cholestasis and focal erythropoiesis. Noteworthy are abnormal numbers and ultrastructural appearance of mitochondria in panel D.
Alpers disease: Example #1 - Valproic acid and acute liver failure in an infant

FIGURE 2. Two cases; Infant #1 (A and B) and infant #2 (C and D). Both examples have intralobular cholestasis, focal inflammation, and mild fatty change. Hepatocyte regeneration is prominent in panel A.
Alpers disease: Example #2 - Complex III defect, valproic acid and acute liver failure in an infant
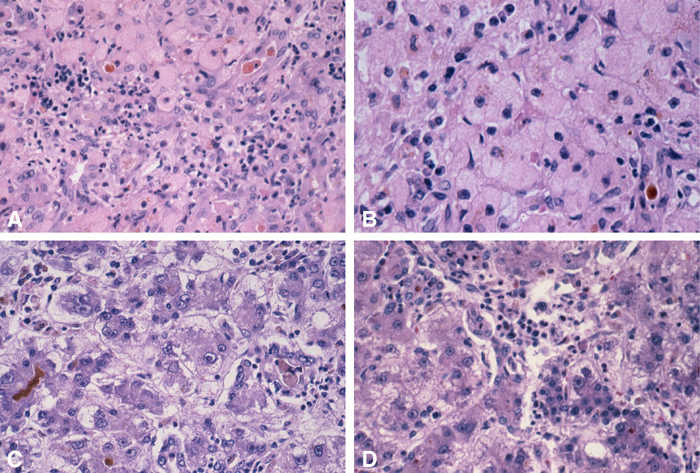
FIGURE 3. Notable features are: A: portal inflammation B and D: microvescicular fat ad mitochondria-laden granular red hepatocytes C: prominent hepatocyte regeneration
Alpers disease: Example #3 - Late stage liver lesion in an infant
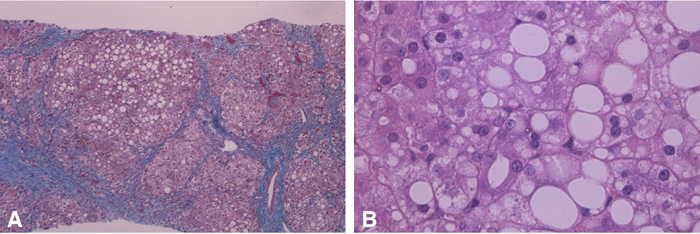
FIGURE 4. A: Micronodular cirrhosis. B: Non-uniform micro and macrovesicular fatty change.
PROGRESSIVE FAMILIAL INTRAHEPATIC CHOLESTASIS
PFIC (primary familial intrahepatic cholestasis). This is an umbrella term for multiple known genetic defects in the complex process of bile secretion from the hepatocytes into the canaliculus. These defects impair transport across the canalicular membrane of critically important constituents of normal bile such as bile acids or phospholipid. Characteristic patterns of liver disease accompany each of these defects which are commonly first suspected upon the basis of liver biopsy findings in conjunction with the clinical and/or family history. Each type is characterized by variable rates of progression and variable histology.
PFIC1
PFIC1 is due to mutation in FIC1 gene ATP8B that encodes an aminoacid phospholipid flipase located on the hepatocyte canalicular membrane. The ability to transport bile constituents is impaired across the canaliculuar membrane, and also across the ileal enterocyte membrane where reabsorbtion normally occurs, producing chronic diarrhea. Bile residue in the canalicular lumen often has a distinctive ultrastuctural appearance that facilitates diagnosis. Hepatic fibrosis is mild to absent.
PFIC1: Liver biopsies at 2 and 8 years in one patient. No fibrosis.
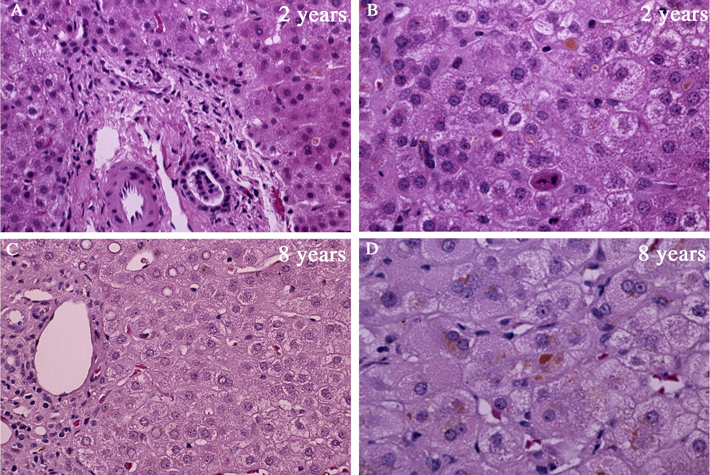
FIGURE 1. A: Age, 2 years. Hepatocytes are normal except for vague rosettes and rare canalicular bile plug. Bile duct in portal area is normal. B: Focal cytoplasmic swelling, rare necrotic hepatocyte C: Age, 8 years. Fibrosis is absent. Trichrome stain. D: Focal cytoplasmic swelling and rare canalicular bile plug
PFIC1: Coarse granular "Byler bile" in a dilated canaliculus, example #1
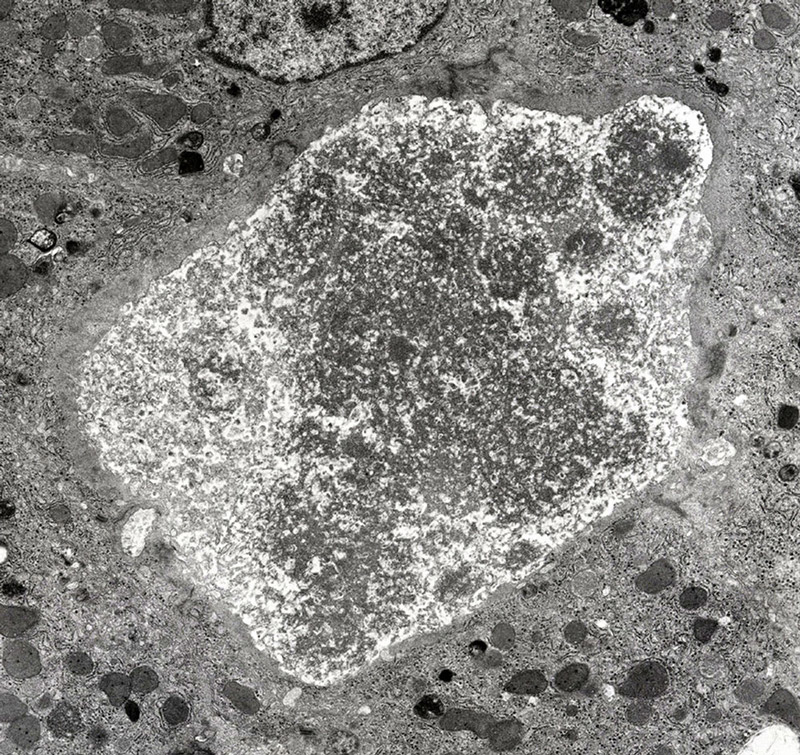
FIGURE 2. Electron Microscopy reveals unusual quality of canalicular bile
PFIC1: Byler bile in canaliculus, example #2
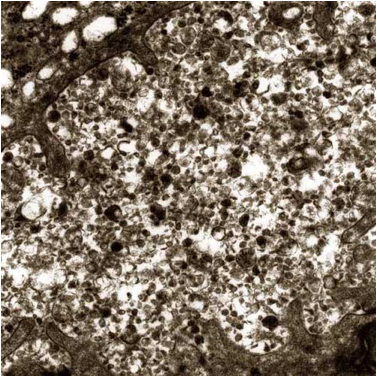
FIGURE 3. Electron Microscopy reveals unusual quality of canalicular bile
PFIC1: Compare canalicular bile in biliary atresia
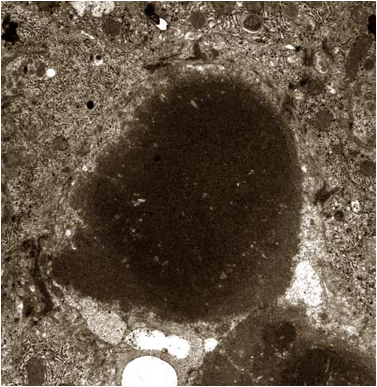
FIGURE 4. Note the difference between PFIC1 bile and ordinary bile in biliary atresia
PFIC1: Compare canalicular bile in estrogen-induced cholestasis
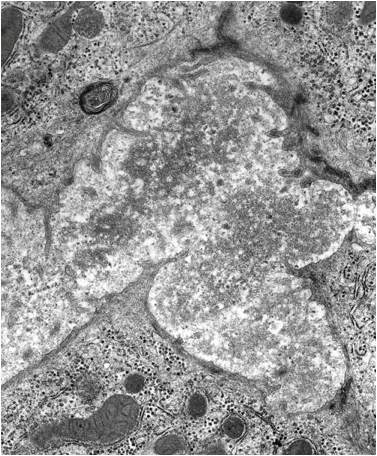
FIGURE 5. Note the difference between PFIC1 late and ordinary bile in estrogen induced cholestasis.
PFIC1: Compare canalicular bile in idiopathic neonatal hepatitis
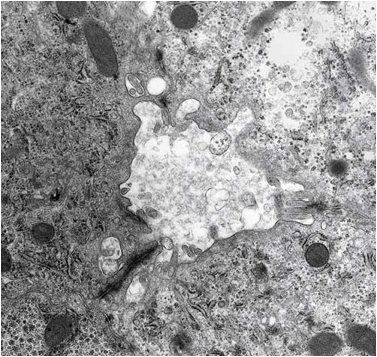
FIGURE 6. Note the difference between PFIC1 late and ordinary bile in idiopathic neonatal hepatitis.
PFIC2
PFIC2 is due to mutations in the gene that codes ABCB11, a transporter of bile acids located in the canalicular membrane know as bile salt export protein (BSEP). Progressive or intermittent cholestasis has extremely varied outcomes.
PFIC2: Non-diagnostic liver biopsy in young infant
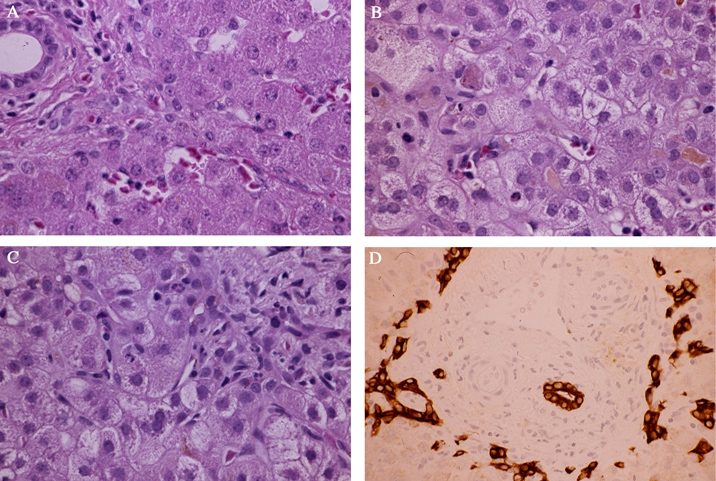
FIGURE 1. A: Normal bile duct and minimal hepatocyte change. B and C: Lobular cholestasis with rare multinucleate giant cell. D: Mild ductular reaction. Cytokeratin immunstain.
PFIC2: Presentation in an infant with rapid progression
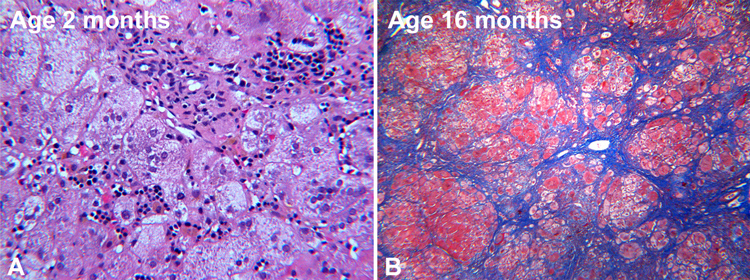
FIGURE 2. A: Age 2 months. Severe lobular cholestasis with prominent giant cell transformation of hepatocytes and extr-medullary hematopoiesis. Interlobular bile ducts are normal. B: Age 16 months. Liver explant. Persistent giant cell transformation with prominent lobular fibrosis. Trichrome stain.
PFIC2: Presentation in an infant with slow progression
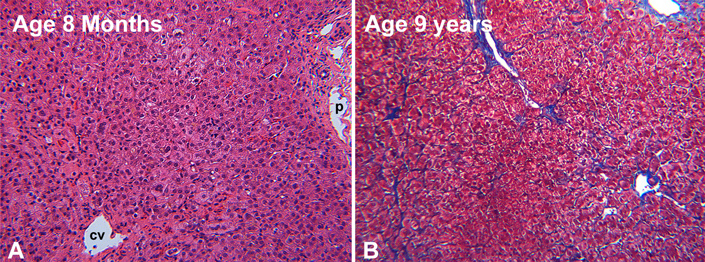
FIGURE 3. A: Age 8 months. Mild lobular cholestasis with scattered small multinucleate hepatocytes and mild pericellular fibrosis. CV,central vein. P, portal zone. B: Age 9 years. No significant change. Minor pericellular and central lobular fibrosis. Trichrome stain.
PFIC3
PFIC3 is due to mutations in the MDR3 gene that codes for ABCB4, a transporter of phospholipid across the canalicular membrane. Absent phospholipid in bile destabilizes bile, unmasking the inherent toxicity of normal bile acids. This results in a disease of the small bile ducts of variable severity and a predilection to develop cholesterol gall stones. Clinical symptoms may be delayed until early adulthood, or pregnancy. A progressive severely destructive cholangiopathy may occur in young children.
PFIC3: Presentation in an infant with profound intrahepatic cholangiopathy and progressive fibrosis
_small.jpg)
FIGURE 1. A: Hepatocyte swelling, giant cell transformation and rare necrotic hepatocyte. B: Prominent ductular reaction at portal margin. C: Proliferation of ducts and ductules simulates changes of mechanical obstruction, Cytokeratin stain. D: Proliferation of ducts associated with periductal fibrosis.
PFIC3: Presentation in late infancy with rapid progression to biliary cirrhosis
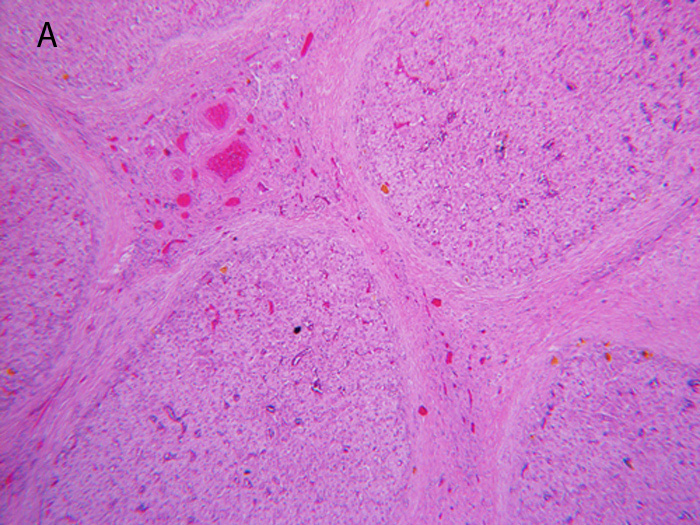
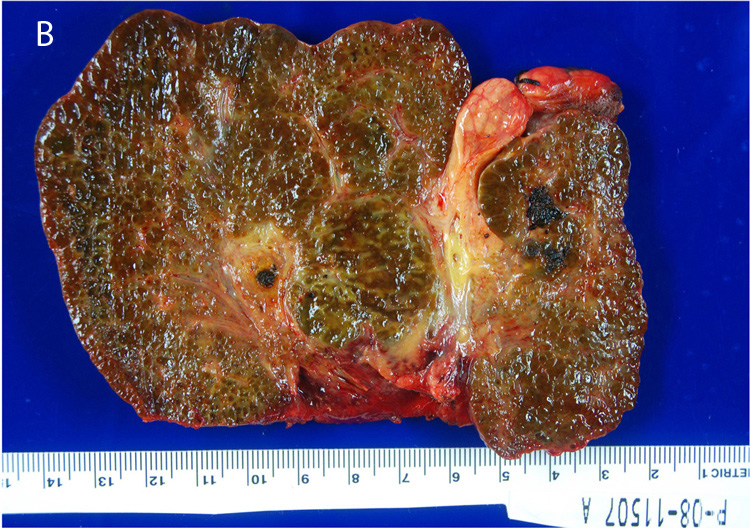
FIGURE 2. A: Cirrhosis with extreme paucity of interlobular bile ducts in explanted liver at age two years. B: Gross photo of explanted liver.